Next Steps in Large-Scale Density Functional Theory
Wednesday, June 24, 2026 3:45 PM to 5:15 PM · 1 hr. 30 min. (Europe/Berlin)
Foyer D-G - 2nd Floor
Research Poster
Chemistry and Materials ScienceMixed PrecisionNovel AlgorithmsParallel Numerical AlgorithmsPhysics
Information
Poster is on display and will be presented at the poster pitch session.
The continued miniaturization of semiconductor devices has pushed characteristic length scales into the nanometer regime, where quantum mechanical effects dominate electronic, chemical, magnetic, and mechanical behavior. Density Functional Theory (DFT) provides a reliable and predictive framework for such systems, but conventional eigenvalue-based formulations scale cubically with system size and become computationally prohibitive for realistic nanoelectronic devices containing millions of atoms. In addition, the global communication patterns inherent to eigensolvers limit scalability on modern massively parallel high-performance computing (HPC) architectures.
Linear-scaling electronic structure methods offer a promising alternative. Density-matrix-based approaches achieve linear scaling by exploiting the nearsightedness of electronic matter, but they rely on a finite band gap and are therefore largely restricted to insulating systems. Metallic systems and semiconductor devices with conducting leads remain challenging. Green’s function-based DFT overcomes this limitation by enabling linear scaling through spatial truncation of long-range interactions while remaining applicable to metallic systems.
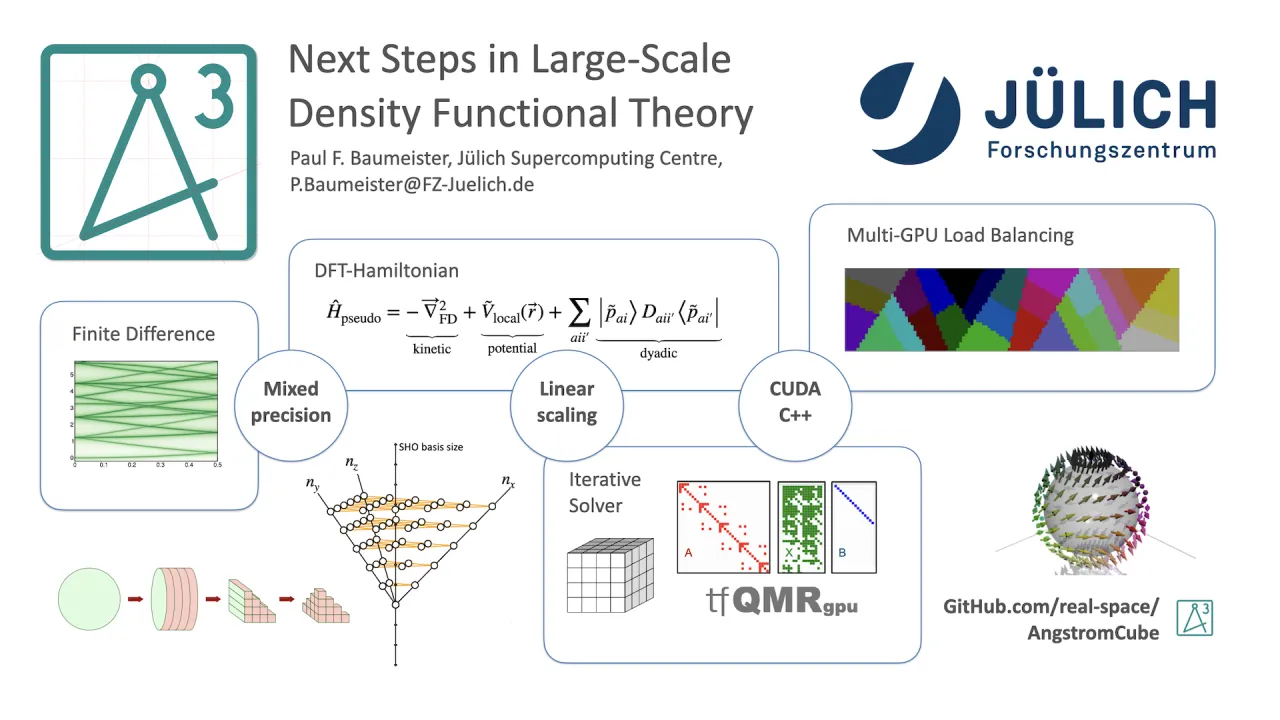
In this work, we present AngstromCube, a real-space Green’s Function Density Functional Theory (RSGF-DFT) application designed explicitly for large-scale GPU-accelerated HPC platforms. Instead of solving the Kohn–Sham eigenvalue problem, AngstromCube computes the time-independent Green’s function of the effective single-particle Hamiltonian. The electron density is obtained from the imaginary part of the diagonal Green’s function via the Kramers–Kronig relation, eliminating explicit band summations. The method requires contour integration in complex energy space, with sampling strategies informed by established techniques from the Korringa–Kohn–Rostoker multiple scattering community.
To achieve plane-wave-level accuracy, all operators are represented on a uniform Cartesian real-space grid using high-order finite-difference stencils, with derivatives up to 16th order supported. Linear scaling is achieved by truncating the Green’s function beyond a finite spatial radius, introducing a second key convergence parameter in addition to the grid spacing. The resulting computational cost exhibits strong sensitivity to both parameters, motivating ongoing work on optimizing the prefactor and identifying crossover points where the linear-scaling approach becomes more efficient than conventional cubic-scaling DFT.
AngstromCube employs the Projector Augmented Wave (PAW) method for electron–ion interactions. A key performance feature is the sparse treatment of non-local projector functions, which are expanded in a factorizable basis of Hermite–Gauss polynomials and evaluated on the fly on each GPU. This minimizes memory bandwidth requirements and avoids storage of large non-local operator matrices.
The core computation is a GPU-accelerated implicit Hamiltonian operator applied iteratively using the transpose-free Quasi-Minimal Residual (tfQMR) method to compute for Green’s function elements. The code is implemented in CUDA-enabled C++ with templated kernels supporting mixed precision, complex arithmetic, and multiple spin formulations. MPI-level parallelization is achieved by distributing independent columns of the truncated Green’s function across MPI tasks with customised load balancing. Mixed-precision strategies are employed to reduce time to solution while maintaining numerical stability.
AngstromCube demonstrates that real-space Green’s function DFT can deliver high physical fidelity together with true linear scaling on modern GPU-accelerated HPC systems, enabling first-principles simulations of nanoelectronic structures and a wide range of other material classes at previously inaccessible scales.
The continued miniaturization of semiconductor devices has pushed characteristic length scales into the nanometer regime, where quantum mechanical effects dominate electronic, chemical, magnetic, and mechanical behavior. Density Functional Theory (DFT) provides a reliable and predictive framework for such systems, but conventional eigenvalue-based formulations scale cubically with system size and become computationally prohibitive for realistic nanoelectronic devices containing millions of atoms. In addition, the global communication patterns inherent to eigensolvers limit scalability on modern massively parallel high-performance computing (HPC) architectures.
Linear-scaling electronic structure methods offer a promising alternative. Density-matrix-based approaches achieve linear scaling by exploiting the nearsightedness of electronic matter, but they rely on a finite band gap and are therefore largely restricted to insulating systems. Metallic systems and semiconductor devices with conducting leads remain challenging. Green’s function-based DFT overcomes this limitation by enabling linear scaling through spatial truncation of long-range interactions while remaining applicable to metallic systems.
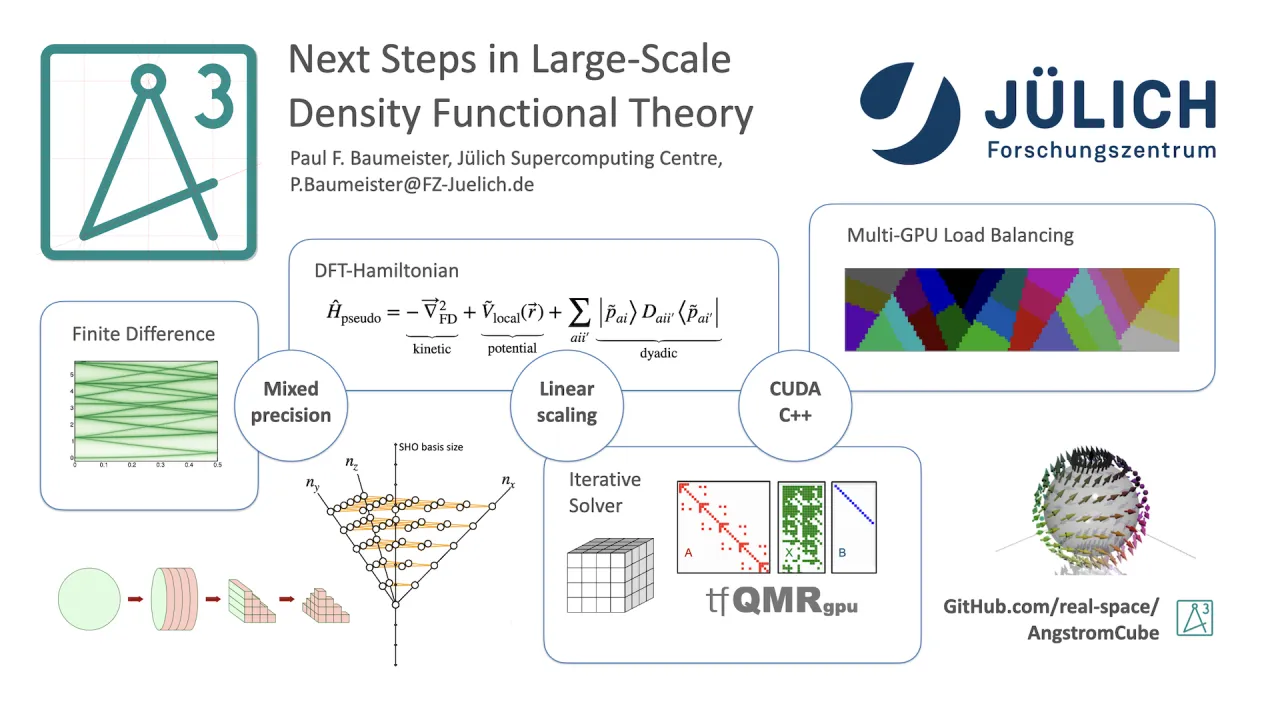
In this work, we present AngstromCube, a real-space Green’s Function Density Functional Theory (RSGF-DFT) application designed explicitly for large-scale GPU-accelerated HPC platforms. Instead of solving the Kohn–Sham eigenvalue problem, AngstromCube computes the time-independent Green’s function of the effective single-particle Hamiltonian. The electron density is obtained from the imaginary part of the diagonal Green’s function via the Kramers–Kronig relation, eliminating explicit band summations. The method requires contour integration in complex energy space, with sampling strategies informed by established techniques from the Korringa–Kohn–Rostoker multiple scattering community.
To achieve plane-wave-level accuracy, all operators are represented on a uniform Cartesian real-space grid using high-order finite-difference stencils, with derivatives up to 16th order supported. Linear scaling is achieved by truncating the Green’s function beyond a finite spatial radius, introducing a second key convergence parameter in addition to the grid spacing. The resulting computational cost exhibits strong sensitivity to both parameters, motivating ongoing work on optimizing the prefactor and identifying crossover points where the linear-scaling approach becomes more efficient than conventional cubic-scaling DFT.
AngstromCube employs the Projector Augmented Wave (PAW) method for electron–ion interactions. A key performance feature is the sparse treatment of non-local projector functions, which are expanded in a factorizable basis of Hermite–Gauss polynomials and evaluated on the fly on each GPU. This minimizes memory bandwidth requirements and avoids storage of large non-local operator matrices.
The core computation is a GPU-accelerated implicit Hamiltonian operator applied iteratively using the transpose-free Quasi-Minimal Residual (tfQMR) method to compute for Green’s function elements. The code is implemented in CUDA-enabled C++ with templated kernels supporting mixed precision, complex arithmetic, and multiple spin formulations. MPI-level parallelization is achieved by distributing independent columns of the truncated Green’s function across MPI tasks with customised load balancing. Mixed-precision strategies are employed to reduce time to solution while maintaining numerical stability.
AngstromCube demonstrates that real-space Green’s function DFT can deliver high physical fidelity together with true linear scaling on modern GPU-accelerated HPC systems, enabling first-principles simulations of nanoelectronic structures and a wide range of other material classes at previously inaccessible scales.
Format
on-demandon-site

